




Lupine Publishers Group
Lupine Publishers
ISSN: 2637-4706
Review Article(ISSN: 2637-4706) 
QSAR and Molecular Docking Studies of Serotonin Derivatives Volume 3 - Issue 1
Received: May 07, 2019; Published: May 13, 2019
Corresponding author: Teodora E Harsa, Faculty of Chemistry and Chemical Engineering, Babes-Bolyai University, Romania
DOI: 10.32474/DDIPIJ.2019.03.000154
Abstract
Serotonin, 5-hydroxytryptamine, represents a class of monoamine neurontransmitters, all of which having a chemical template comprised of a basic amino group separated from an aromatic nucleus by a two-carbon aliphatic chain. In this paper we present a docking study on serotonin targeting the proteins 3ADX and 2YX8, respectively, performed by AutoDock Vina. A set of thirtyfive serotonins, downloaded from PubChem, was modeled, within the hypermolecule strategy; the predicted activity was LD50 and prediction was done on similarity clusters with the leaders chosen as the best docked ligands on the Peroxisome proliferatoractivated receptor gamma. It was concluded that LD50 of the studied serotonins is not directly influenced by their binding energies to the target proteins.
Keywords: Serotonin; Docking; Binding affinity; 3ADX; 2YX8; Hypermolecule; LD50; QSAR
Introduction
Serotonin is a neurotransmitter involved in a wide variety of behaviors, including feeding and body-weight regulation, social hierarchies, aggression and suicidality, obsessive compulsive disorder, alcoholism, anxiety, and affective disorders [1-3]. Peroxisome proliferator-activated receptors are ligandactivated transcription factors that regulate genes important in cell differentiation and various metabolic processes, especially lipid and glucose homeostasis [4].
Receptor activity-modifying proteins (RAMPs) are a class of proteins that interact with and modulate the activities of several Class B G Protein-Coupled Receptors including the receptors for secretin, calcitonin (CT) and glucagon, RAMPs complex with Sumatriptan molecule [5].
A method is described to dock a serotonin into a binding site in a peroxisome proliferator-activated receptor gamma on the basis of the complementarity of the inter-molecular atomic contacts. Docking is performed by maximization of a complementarity function that is dependent on atomic contact surface area and the chemical properties of the contacting atoms [6].
Serotonin is metabolized in cells of the brain, gastrointestinal tract, liver, lungs and platelets; it exerts its effect on endothelial cells through multiple receptors [7].
A pharmacophore model can be established either in a ligand based manner, by superposing a set of active molecules with low binding energy [8] and extracting common chemical features that are essential for their bioactivity, or in a structure-based manner, by probing possible interaction points between the protein target and serotonins [9,10].
In our previous work [11], we performed a QSAR study on a set of serotonins, by the similarity cluster prediction approach, proposed by TOPO Group Cluj. In this paper, a docking study, performed to identify the geometric description of the pharmacophore in the interaction of this class of ligands with the peroxisome proliferatoractivated receptor gamma, is reported. We developed a new approach by creating similarity clusters using as leaders those ligands showing the best scores in the docking test.
Docking Study
A set of 35 derivatives of serotonin were taken from PubChem Database [12] and were divided into a training set (25 molecules) and a test set (10 molecules, in Italics), taken with the lowest docking energy (Table 1). The property chosen for modeling was LD50 (on rat, intraperitoneal route administered).
The molecular docking study was carried out to explore the binding mode of serotonin derivatives (Table 2) within the binding energy of peroxisome proliferator-activated receptor gamma 3ADX and Receptor activity-modifying protein 1 2YX8.
Table 1: Serotonin molecular structures (in SMILES code) and LD50 (taken from PubChem).

Table 2: List of Molar Mass, Molecular Formulas and Torsions of the Ligands.

Figure 1:The proteins (Peroxisome proliferator-activated receptor gamma left, Receptor activity-modifying protein 1 right) (RCSB PDB CODE: 3ADX, 2YX8).

Molecular graphics laboratory (MGL) tools and AutoDock4.2 was downloaded from www.scripps.edu [14]. We employed the Lamarckian genetic algorithm (LGA) for ligand conformational searching [15]. AutoDock Tools was used for creating PDBQT files from traditional PDB files. The grid menu is toggled [16]; after loading protein. pdbqt the map files were selected directly with setting up the grid points appropriate for the searching of ligand within the active site of the protein molecule. This way the grid parameter files are created with setting up the map files directly. The docking parameter files were completed by using the Lamarckian genetic algorithm [17].
(Figure 2) illustrates the final Lamarckian genetic algorithm docked state: binding energy of ligands with the active site of Peroxisome proliferator-activated receptor gamma (3ADX). Docking energies lie in the range: -7.0 and -5.1kcal/mol. It seems that the higher the number of rotatable bonds, the higher the ligand–enzyme affinity is.
The distribution of the docking energy (Figure 2) may be attributed to the differences in position of the functional groups within the studied compounds. Interaction of the ligand with the protein can be seen in (Figure 3).
Figure 2: Binding free energies for the interaction of serotonins with 3ADX target.

Figure 3: The interaction of Serotonin with peroxisome proliferator-activated receptor gamma.

(Figure 4) provides the docking data for the interaction of serotonins with the active site of Receptor activity-modifying protein 1 (2YX8): the lowest binding energy was -6.0 while the highest, -4.9kcal/mol.
Figure 4: Binding free energies for the interaction of serotonins with 2YX8 target.

To build the pharmacophore model for the serotonins interacting with the peroxisome proliferator-activated receptor gamma, the conformers showing the best scores in docking where chosen. Ligands 6, 20, 32 and 33 have the lowest binding energy, between -7 and -6.8; based on these compounds, the pharmacophore was drawn up [18] (Figure 5). This was achieved with HyperChem7.52 [19] and PyMOL [20] software.
Figure 5(a): Pharmacophore model for the Serotonins interacting with peroxisome proliferator-activated receptor gamma.

Figure 5(b): Pharmacophore model for the Serotonins interacting with peroxisome proliferator-activated receptor gamma.

Pharmacophore comprises the following features:
a. hydrophobic interaction and π-electron delocalized indole ring of serotonin bearing a nitrogen atom
b. hydrogen bond acceptor centre located on the oxo group
c. hydrogen bond donor centre located on 5-hydroxyl group
d. hydrogen bond donor center located on 3-acetate group
Computational
A set of 35 molecular structures, belonging to the class of Serotonin, have been downloaded from the Pubchem database (Table 1), together with their LD50. The set was split into the training set and test set (25 and 10 molecules, respectively), the test set including the molecules with the lowest docking energy (Table 3). A hypermolecule (Figure 6) was built up as the reunion of all structural features in the molecules under study. The structures have been optimized at HF (3-21g(,p)) level of theory, in gas phase, by Gaussian 09 [21]. Topological indices have been computed by TOPOCLUJ software [22]; some of them (Cluj indices, SD) and electronic parameters (HOMO, LUMO, electrophilicity index, ω, and chemical strength, π) are listed in Table 3.
Figure 6: Hypermolecule.

Table 3: LD50, SD, topological and electronic indices for 35 serotonins (see Table 1).

The electrophilicity index, ω, is calculated as:
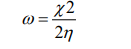
with 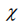 being the electronegativity and
being the electronegativity and 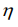 chemical hardness.
chemical hardness.
Chemical strength, π, quantifies the activation energy necessary to interchange the maximum number of electrons in a chemical reaction [23].
QSAR Study
This QSAR study was performed following Diudea’s algorithm [24]; it is based on the alignment of molecules over a hypermolecule [25] and a correlation weighting procedure [26,27] coupled with a predictive validation of the model descriptors within similarity clusters [28] performed for each molecule in the test set.
Partial charges description (LD50)
Before performing the models, the descriptors with variance
Var<10% and intercorrelation larger than 0.80 were discarded.
Next, correlation weighting was performed on all the positions in
the hypermolecule: the correlating coefficients of the statistically
significant positions of the hypermolecule were used to multiply
the local descriptors, actually the partial charges, thus resulting
new weighted vectors  . Then, the local correlating
descriptors are summed to give a global descriptor, i ij j
. Then, the local correlating
descriptors are summed to give a global descriptor, i ij j 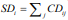 This new descriptor is a linear combination of the local correlating
descriptors for the significant positions in the hypermolecule: H2,
H3, H4, H6, H8, H10, H11, H14, H15, H16, H17, H19, H20, H21, H22,
H24). The resulting SD global descriptor will be used as the basis
for modeling LD50 (see Table 1). SD correlates with LD50 as below:
This new descriptor is a linear combination of the local correlating
descriptors for the significant positions in the hypermolecule: H2,
H3, H4, H6, H8, H10, H11, H14, H15, H16, H17, H19, H20, H21, H22,
H24). The resulting SD global descriptor will be used as the basis
for modeling LD50 (see Table 1). SD correlates with LD50 as below:
LD50= SD-428.792, N=35; R2=0.8521; s=130.602; F = 151.213
QSAR models
The models were performed on the training set (25 structures in Table 1) and the best results (in decreasing order of R2 ) are listed below and in (Table 4); the test set collects the molecules with the lowest docking energies.
(i) Monovariate regression
LD50 = -414.965+ 0.971×SD
(ii) Bivariate regression
LD50 = -611.96 + 0.956×SD +11.261×C
(iii) Three-variate regression
LD50 = -354.713+ 0.939×SD-1.191×CjDi+2.031×CjDe
Model validation
Leave-one-out: The performances in leave-one-out analysis [29] related to the models listed as best in (Table 4) are presented in (Table 5).
Table 4: The best models in LD50 in the training set of serotonin in Table 1.

Table 5: The best models in LD50 in the training set of serotonin in Table 1.

External validation: The values LD50 for the test set of
serotonin were calculated by using equation 11 in (Table 4).
Data are listed in (Table 6) and the monovariate correlation:
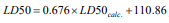 ; n=10, R2=0.742; s=182.028;
F=22.983 is plotted in (Figure 7).
; n=10, R2=0.742; s=182.028;
F=22.983 is plotted in (Figure 7).
Table 6: The best models in LD50 in the training set of serotonin in Table 1.

Figure 7: The plot LD50 vs. LD50calc. for the test set (external validation).

Similarity cluster validation: The values LD50 for the test
set of serotonin were calculated by using equation 11 in (Table
4). Data are listed in (Table 7) and the monovariate correlation:
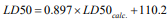 ; n=10; R2=0.962; s=69.361;
F=205.390 is plotted in (Figure 8) [30,31].
; n=10; R2=0.962; s=69.361;
F=205.390 is plotted in (Figure 8) [30,31].
Table 7: Calculated values of LD50 by similarity clusters, for the molecules in the test set.

Figure 8: The plot LD50 vs. LD50calc. for the test set (external validation).

Prediction of LD50 is more accurate when using the similarity clusters (R2= 0.962), compared to the classical external validation of the model (R2= 0.742). The binding energies provided by the docking step correlate very poorly (R2= 0.009 for 3ADX and R2= 0.242 for 2YX8 with the LD50 values); however, selection of the leaders in the similarity test as the best docked ligands clearly improved the LD50 prediction. The binding energies correlate better (n=35; R2=0.614; for 3ADX with LUMO energies and R2=0.626; for 2YX8 with CjDe). On the presented data, we cannot say clearly that LD50 of the studied serotonins is determined by the interaction of these ligands with the two target proteins; further studies are however needed for the elucidation of this question.
Conclusion
In this article, a docking study on 35 serotonin derivatives, over Peroxisome proliferator-activated receptor gamma (3ADX) and Receptor activity-modifying protein 1(2YX8), was performed. The initial set of molecules was split into a training set and the test set (chosen as the best scoring ligand in the docking test on protein 3ADX); a QSAR model was performed on 25 of these structures, with respect to their LD50. By structure similarity clustering, we ensured a “congeneric state” within the set of molecules used next to predict the LD50 in the test set; by this procedure, the accuracy of prediction surpassed the model (R2=0.962 in prediction vs. 0.864 in the model). It was concluded that LD50 of the studied serotonins is not directly influenced by their binding energies to the target proteins.
Acknowledgements
This paper is a result of a doctoral research made possible by the financial support of the Sectoral Operational Programme for Human Resources Development 2007-2013, co-financed by the European Social Fund, under the project POSDRU/159/1.5/S/137750 - “Doctoral and postoctoral programs - support for increasing research competitiveness in the field of exact Sciences”.
References
- Brodfuehrer PR, Chen BC, Sattelberg TR, Smith PR, Reddy JP, et al. (1997) An Efficient Fischer Indole Synthesis of Avitriptan, a Potent 5-HT1D Receptor Agonist. J Org Chem 62: 9192-9202.
- Best JA, Nijhout HF, Reed M (2010) Serotonin synthesis, release and reuptake in terminals: a mathematical model. Theoretical Biology and Medical Modelling 19(7): 34.
- Tepper SJ, Rapoport AM, Sheftell FD (2002) Mechanisms of action of the 5-HT1B/1D receptor agonists. Arch Neurol 59(7): 1084-1088.
- Schaffer JE (2003) PPAR-gamma has been implicated in the pathology of numerous diseases including obesity, diabetes, atherosclerosis, and cancer. Lipotoxicity: when tissues overeat. Curr Opin Lipidol 14: 281- 287.
- Zhang Z, Winborn CS, Marquez de Prado B, Russo AF (2007) Sensitization of Calcitonin Gene-Related Peptide Receptors by Receptor Activity-Modifying Protein-1 in the Trigeminal Ganglion. The Journal of Neuroscience 27(10): 2693-2703.
- Berger J, Moller DE (2002) The mechanisms of action of PPARs. Annu Rev Med 53: 409-435.
- Sobolev V, Wade RC, Vriend G, Edelman M (1996) Molecular docking using surface complementarity. PROTEINS: Structure Function and Genetics 25: 120-129.
- Dror O, Alexandra Shulman-Peleg, Ruth Nussinov, Haim J Wolfson (2006) Predicting molecular interactions in silico. I. An updated guide to pharmacophore identification and its applications to drug design. Front Med Chem 3: 551-584.
- Schloss P, Williams DC (1998) The serotonin transporter: a primary target for antidepressant drugs. J Psychopharmacol 12(2): 115-121.
- Wermuth CG (2006) Pharmacophores: historical perspective and viewpoint from a medicinal chemist. In: Langer T, Hoffmann RD [Edt.], Pharmacophores and Pharmacophore Searches. Wiley-VCH, pp. 3-13.
- Harsa TE, Harsa AM, Jantschi L, Diudea MV (2015) QSAR study on Indole derivatives. Journal of Chemical and Pharmaceutical Research 7(3): 2378-2393.
- PubChem database, accessed 23.06. 2015.
- The RCBS Protein data bank.
- Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem 31(2): 455-461.
- Khodade P, Prabhu R, Chandra N (2007) Parallel implementation of Autodock. J App Crystal 40: 598-599.
- Dhananjayan K, Kalathil K, Sumathy A, Sivanandy P (2014) A computational study on binding affinity of Bio-flavonoids on the crystal structure of 3-hydroxy-3-methyl-glutaryl-CoA reductase - An insilico molecular docking approach. Der Pharma Chemica 6(2): 378-387.
- Abagyan R, Totrov M (2001) High-throughput docking for lead generation. Curr Opin Chem Biol 5(4): 375-82.
- Kołaczkowski M, Nowak M, Pawłowski M, Bojarski AJ (2006) ReceptorBased Pharmacophores for Serotonin 5-HT7R Antagonists Implications to Selectivity. Journal of Medicinal Chemistry 49(23): 6732-6741.
- HyperChem 7.52, Hypercube, Inc.
- PyMol 0.97, DeLano Scientific.
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA (2009) Gaussian 09. Fox, Gaussian Inc Wallingford CT.
- Ursu O, Diudea MV (2005) TOPOCLUJ software program. Babes-Bolyai University, Cluj, Romania.
- Putz MV, Tudoran AM, Putz AM (2013) Structure-Properties and Chemical-Bio/Ecological Interactions of PAHs: from Synthesis to Cosmic Spectral Lines and Nano-chemistry, to Reactivity Driving Lipophilicity. Current Organic Chemistry 17(23): 2845-2871.
- Moldovan CD, Costescu A, Katona G, Diudea MV (2008) A novel QSAR approach in modeling antifungal activity of some 5- or 6-methyl-2- substituded benzoxazoles/ benzimidazoles against C. albicans using molecular descriptors. MATCH Commun Math Comput Chem 60: 977- 984.
- Balaban AT, Chiriac A, Motoc I, Simon Z (1980) Steric Fit in QSAR. Lectures Notes in Chemistry, Springer.
- Toropov AA, Toropova AP (2002) QSAR modeling of mutagenicity based on graphs of atomic orbitals. Internet El J Molec Design 1: 108-114
- Toropov AA, Toropova AP (2001) Prediction of heteroaromatic amine mutagenicity by means of correlation weighting of atomic orbital graph of local invariants. J Mol Struct (Theochem) 538: 287-293.
- Willett P (2013) Combinations of similarity rankings using data fusion. J Chem Inf Model 53: 1-10.
- Jäntschi L (2005) LOO Analysis (LOO: leave one out). Academic Direct Library of software.
- Harsa TE, Harsa AM, Szefler B (2014) QSAR of Caffeines by similarity cluster prediction. Cent Eur J Chem 12: 365-376.
- Harsa AM, Harsa TE, Bolboaca S, Diudea MV (2014) QSAR in flavonoids by similarity cluster prediction. Curr Comput Aided Drug Design 10: 115-128.



-

Mark E Smith
Bio chemistry
University of Texas Medical Branch, USA -

Lawrence A Presley
Department of Criminal Justice
Liberty University, USA -

Thomas W Miller
Department of Psychiatry
University of Kentucky, USA -

Gjumrakch Aliev
Department of Medicine
Gally International Biomedical Research & Consulting LLC, USA -

Christopher Bryant
Department of Urbanisation and Agricultural
Montreal university, USA -

Robert William Frare
Oral & Maxillofacial Pathology
New York University, USA -

Rudolph Modesto Navari
Gastroenterology and Hepatology
University of Alabama, UK -

Andrew Hague
Department of Medicine
Universities of Bradford, UK -

George Gregory Buttigieg
Maltese College of Obstetrics and Gynaecology, Europe -

Chen-Hsiung Yeh
Oncology
Circulogene Theranostics, England -
.png)
Emilio Bucio-Carrillo
Radiation Chemistry
National University of Mexico, USA -
.jpg)
Casey J Grenier
Analytical Chemistry
Wentworth Institute of Technology, USA -

Hany Atalah
Minimally Invasive Surgery
Mercer University school of Medicine, USA -

Abu-Hussein Muhamad
Pediatric Dentistry
University of Athens , Greece
