



Lupine Publishers Group
Lupine Publishers
ISSN: 2638-5945
Review Article(ISSN: 2638-5945) 
Somatic Mutations in Cancer-Free Individuals: A Liquid Biopsy Connection Volume 1 - Issue 1
Received: December 19, 2017; Published: January 18, 2018
*Corresponding author: Chen Hsiung Yeh, PhD Circulogene Theranostics 3125 Independence Dr, Suite 301 Birmingham, AL 35209, USA
DOI: 10.32474/OAJOM.2018.01.000101
Also view in:




Abstract
Somatic mutations have been perceived as the causal event in the origin of the vast majority of cancers. Advanced massively parallel, highthroughput DNA sequencing have enabled the comprehensive characterization of somatic mutations in a large number of tumor samples for precision and personalized therapy. Understanding how these observed genetic alterations give rise to specific cancer phenotypes represents an ultimate goal of cancer genomics. However, somatic mutations are also commonly found in healthy individuals, which interfere with the effectiveness for cancer diagnostics.
Keywords: Somatic mutation; Germline; Cell-free DNA; Liquid biopsy; Next-generation sequencing
Abbreviations: NGS: Next-Generation Sequencing ; cfDNA: Cell-free DNA; MAF: Mutant Allele Frequency
Introduction
Mutations in healthy individuals are not all germline
Over the course of our lifetime, there are many millions of cell divisions in the body. By chance alone, mutations will definitely occur. Indeed, spontaneous somatic mutations constantly occur in individual cells. These background mutations arise either from replication errors or from DNA damage that is repaired incorrectly or left unrepaired, and have been detected in healthy tissues, including blood, skin, liver, colon, and small intestine [1-3]. Deepsequencing studies in normal tissues also surprisingly identified cancer-driving mutations, e.g., in blood, driver mutations can be detected in ~10%of individuals older than 65 years of age and resemble patterns seen in leukemia patients. Individuals carrying these driver mutations have an elevated future risk of blood cancers [4-6], suggesting that these are genuine precancerous clones. Further, a detailed analysis of 31,717 cancer cases and 26,136 cancer-free controls from 13 genome-wide association studies revealed that the majority, if not all, of aberrations that were observed in the cancer-associated cohort were also seen in cancer-free subjects, albeit at lower frequency [7,8].
Somatic mutations in healthy individuals are very prevalent, with an average mutation number of around 2–6 mutations/1 M bases [9,10]. The baseline somatic mutation spectrum in healthy population not only can help fill the gaps for the establishing early cancer diagnosis strategies, but also argues against the idea of using normal cells as germline control to make somatic mutation calls in sequencing tests. Moreover, the same driver mutation could exist in both tumor and normal cells yet with distinct biological effects, we should not simply define the threshold of mutation detection by removing the background mutations found in a healthy population. Taken together, we need to incorporate and carefully calibrate the background somatic mutations in healthy individuals; the fact is they are not all germline mutations.
Somatic driver mutations found in healthy population by liquid biopsy
With the dramatically decreased cost of next-generation sequencing (NGS) in recent years, it is now practical to screen a large number of individuals at ultra-deep sequencing depths to identify cancer-related mutations. Cell-free DNA (cfDNA) in the blood circulation of cancer patients (as liquid biopsy) have emerged as key biomarkers for cancer monitoring and treatment decisionmaking [11]. Both academic research groups and industry players are chasing the pan-cancer screening by a simple blood draw. However, the reliable and accurate application of cfDNA detection requires better understanding of background somatic information in healthy individuals.
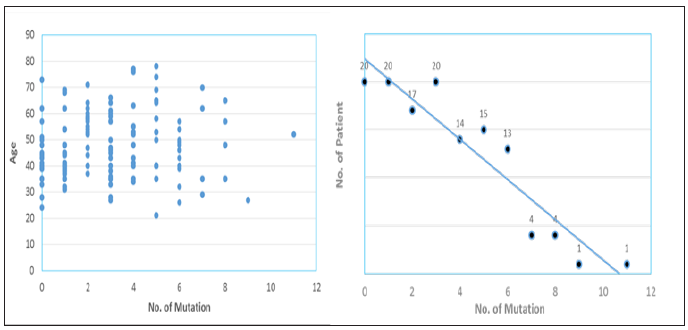
We performed ultra-deep target sequencing on 50 cancerassociated genes for plasma cfDNA from a cohort of 129 apparently healthy cancer-free subjects. To increase the confidence of the called mutations, we here defined the mutation as the variant allele frequency greater than 1% and the average depth more than 5,000 xs for demonstration. Our data revealed an age-independent mutation spectrum with average 3.12 somatic mutations per subject (Figure 1). The most frequently mutated genes are TP53 (42%), KIT (6%), KDR (5.5%), PIK3CA (5.5%), EGFR (5%) and PTEN (3.7%). These results highlighted the prevalence of some cancer-associated driver mutations in healthy individuals as background mutations. We also demonstrated the concordance between our results and a recent study for revealing the real somatic mutation in healthy population.
Figure 1: Distribution plots of somatic mutation detected in a cohort of 129 healthy subjects.
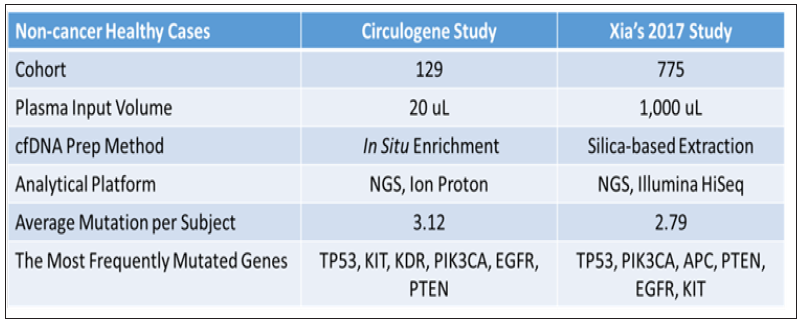
The study by Xia et al. [12] examined the background somatic mutations in white blood cells and cfDNA in healthy controls based on sequencing data from 821 non-cancer individuals with the aim of understanding the baseline profile of somatic mutations detected in cfDNA. The data comparison was summarized in Figure 2. Although there are differences in study cohort composition, sample volume, extraction methodology and analytical platform, the end results are remarkably similar, i.e., average 3 mutations per subject with an almost identical list of frequently mutated genes. Although varying mutation spectra in cancers have often been attributed to cancerspecific processes, our data suggest that at least a subset of these mutations actually reflect normal tissue-specific processes. This concept is consistent with the idea that a substantial fraction of the mutations found in cancers occur in normal stem cells [13,14].
Figure 2: Comparison of somatic mutation detection in healthy population from two studies.
Normal tissue as a germline control not justified
There is evidence for the presence of tumor-derived cfDNA in early cancers [15]. However, the real fraction of cfDNA that shed by tumor rather than the background somatic mutations is not well illustrated. For clinical application, the low level of tumor mutation as well as the heterogeneity of background mutation present in the circulation needs to be clearly addressed and differentiated to achieve accuracy. Unfortunately, this goal can’t be achieved by pushing detection limit of current advanced technology to below 0.01% mutant allele frequency (MAF). Contrarily, the higher sensitivity will guarantee higher chance to pick up background somatic mutations. Also, the clinical relevance of those lowpercentage tumor mutations is still debatable in terms of treatment decision or regimen change. Each human individual is unique. Every cancer patient is different. No two tumors are the same even resides within the same patient; to distinguish the definitive cancer-specific mutations from background signals observable in plasma is extremely daunting. Evaluation of specificity in plasma cfDNA profiles from large numbers of healthy individuals as representative controls for the cancer population seems farfetched with uncertainty, especially when standardized protocol and optimized technology are still lacking.
Unlike tissue genomic DNA, circulating cfDNA is so diluted and dynamic with a relatively short half-life, making single-point measurement not suitable for clinical application. We reason that cfDNA in circulation is truly under a continuous selection pressure to select for highly aggressive/proliferative clones, as disease progressing the low-abundant tumor clones will either evolve and dominate or vanish by the immune clean-up processes, therefore longitudinal clinical follow-up should be performed to identify the best time and target for precision therapy, meanwhile to filter out contaminating background mutations. To achieve high clinical specificity, a cfDNA-based test must be capable of distinguishing between the background signals originating from non-cancer or pre-cancerous processes and the invasive malignancy of clinical interest. It is still possible that mutational signatures in cfDNA could distinguish basic biological processes from malignant and pathological processes.
Figure 3: A representative mutational trending curve after filtering out background mutations.
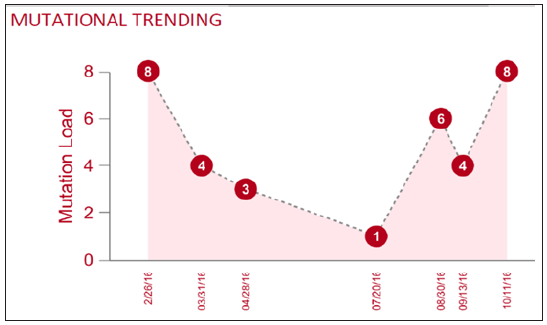
Here we propose a combined approach based on the tumor evolutional principle of “survival and domination of the fittest” in circulation that is to perform multiple time-point monitoring, filter out potential background mutations (e.g., <1% MAF), reduce sample input volume and interrogate multiple databases. A representative mutational trending curve following our approaches was shown (Figure 3). Our findings underscore the importance of an assessment of the landscape of somatic mutations in cancerfree population, and associated mutation signatures. Somatic mutations and mosaicism in healthy individuals have implications not only for early detection, diagnosis and treatment of cancer using liquid biopsy but also emerging technologies in healthcare. We recommend caution while extending the mutation conclusions to cancer patients by employing matched normal tissue as germline control. To increase sample input and push liquid biopsy sensitivity toward <1% may not serve the interest of detecting low-frequency mutant allele, but only to increase the chance of background mutation contamination. Application of artificial intelligence, machine-learning on big database to create an algorithm for highrisk population screening of cancer is a good idea for preventive medicine, yet the outcome is uncertain given the uniqueness of every patient, each tumor - one size can’t fit all.
References
- Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, et al. (2016) Tissuespecific mutation accumulation in human adult stem cells during life. Nature 538: 260-264.
- Martincorena I, Campbell PJ (2015) Somatic mutation in cancer and normal cells. Science 349: 1483-1489.
- Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, et al. (2012) the origin and evolution of mutations in acute myeloid leukemia. Cell 150: 264- 278.
- Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, et al. (2014) Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 371: 2477-2487.
- Jaiswal S, Fontanillas P, Flannick J, ManningA, Grauman PV, et al. (2014) Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371: 2488-2498.
- McKerrell T, Park N, Moreno T, Grove CS, Ponstingl H, et al. (2015) Leukemia-associated somatic mutations drive distinct patterns of agerelated clonal hemopoiesis. Cell Rep 10: 1239-1245.
- Jacobs KB, Yeager M, Zhou W, Wacholder S, Wang Z, et al. (2012) Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet 44: 651-658.
- Forsberg LA, Absher D, Dumanski JP (2013) Non-heritable genetics of human disease: spotlight on post-zygotic genetic variation acquired during lifetime. J Med Genet 50(1): 1-10.
- Volik S, Alcaide M, Morin RD, Collins C (2016) Cell-free DNA (cfDNA): Clinical significance and utility in cancer shaped by emerging technologies. Mol Cancer Res 14: 898-908.
- Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, et al. (2015) Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348: 880-886.
- Lo YM, Corbetta N, Chamberlain PF, Rai V, Sargent IL, et al. (1997) Presence of fetal DNA in maternal plasma and serum. Lancet 350: 485- 487.
- Xia L, Li Z, Zhou B, Tian G, Zeng L, et al. (2017) Statistical analysis of mutant allele frequency level of circulating cell-free DNA and blood cells in healthy individuals. Sci Rep 7: 7526.
- Tomasetti C, Vogelstein B, Parmigiani G (2013) half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc Natl Acad Sci USA 110(6): 1999-2004.
- Tomasetti C, Vogelstein B (2015) Cancer etiology: Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347: 78-81.
- Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, et al. (2014) Detection of circulating tumor DNA in early- and late-stage human malignancies. SciTransl Med 6: 224ra24.


-

Mark E Smith
Bio chemistry
University of Texas Medical Branch, USA -

Lawrence A Presley
Department of Criminal Justice
Liberty University, USA -

Thomas W Miller
Department of Psychiatry
University of Kentucky, USA -

Gjumrakch Aliev
Department of Medicine
Gally International Biomedical Research & Consulting LLC, USA -

Christopher Bryant
Department of Urbanisation and Agricultural
Montreal university, USA -

Robert William Frare
Oral & Maxillofacial Pathology
New York University, USA -

Rudolph Modesto Navari
Gastroenterology and Hepatology
University of Alabama, UK -

Andrew Hague
Department of Medicine
Universities of Bradford, UK -

George Gregory Buttigieg
Maltese College of Obstetrics and Gynaecology, Europe -

Chen-Hsiung Yeh
Oncology
Circulogene Theranostics, England -
.png)
Emilio Bucio-Carrillo
Radiation Chemistry
National University of Mexico, USA -
.jpg)
Casey J Grenier
Analytical Chemistry
Wentworth Institute of Technology, USA -

Hany Atalah
Minimally Invasive Surgery
Mercer University school of Medicine, USA -

Abu-Hussein Muhamad
Pediatric Dentistry
University of Athens , Greece
