




Lupine Publishers Group
Lupine Publishers
ISSN: 2637-4579
Research Article(ISSN: 2637-4579) 
Thermodynamic, HOMO-LUMO, MEP and ADMET Studies of Metronidazole and its Modified Derivatives Based on DFT Volume 3 - Issue 1
Received: January 28, 2018; Published: February 04, 2019
*Corresponding author: Moniruzzaman, Department of Applied Chemistry and Biochemical Engineering, Shizuoka University, 3-5-1 Johoku, Hamamatsu, Shizuoka 432-8561, Japan
DOI: 10.32474/OAJBEB.2019.03.000153
abstract
In this study, Metronidazole (Met) and it’s modified derivatives are optimized by employing density functional theory with B3LYP/6-31g (d,p) level theory to explore their structural and thermodynamical properties. Molecular electrostatic potential (MEP) calculation has performed to calculate their possible electrophilic and nucleophilic attack. ADMET prediction was performed to search the absorption, metabolism and toxic level. Finally, this study can be helpful to design a potent candidate.
Keywords: Metronidazole; Density functional theory; HOMO-LUMO; MEP; ADMET
Abbrevations:Met: Metronidazole; DFT: Density functional theory; HOMO: Highest occupied molecular orbital; LUMO: Lowest unoccupied molecular orbital; MEP: Molecular electrostatic potential; ADMET: Absorption, distribution, metabolism, excretion, and toxicity
Introduction
Metronidazole (Met) is an antibiotic [1] and antiprotozoal drug [2]. It is widely used in the treatment of amoebiasis, trichomoniasis and giardiasis [3,4]. It has some demerits depending on the type and nature of unusual physical condition and on the limit of dose. High and long term dose can cause of leucopenia, neutropenia and peripheral neuropathy diseases [5]. Adverse effect and resistance of drugs indicate the importance of the discovery of new potential candidate. In computer aided drug design system, physicochemical, molecular docking, nonbonding interactions, and ADMET predictions are important criteria to evaluate newly designed molecules [6]. Drug modification is another alternative way to search better agent, which can increase the selective action of drug and reduce the side effect. Recently, it has been seen the trait of modifying drugs using halogens and alkyl group play important role in improving drug performance [7].
Herein, I report the optimization of Metronidazole (Met) and its modified derivatives to investigate their biochemical behavior on the basis of quantum mechanical approach. The free energy, electronic energy, enthalpy, dipole moment, HOMO-LUMO gap, hardness, softness, chemical potential and electrostatic potential have been calculated. All the newly designed derivatives show better thermodynamic properties, and some of them exhibit better chemical reactivity than parent drug. From the regarding quantum chemical studies, it’s assuming that, some of the designed compounds may have profound effect as drug.
Methods and Materials
Computational Details
In computer aided drug design, quantum mechanical methods are widely used to predict thermal, molecular orbital, and molecular electrostatic potential properties [8]. Initial geometry of Metronidazole (Met) was taken from the online structure database named ChemSpider [9]. Geometry optimization and further modification of all structures carried out using Gaussian 09 program [10]. Density functional theory (DFT) with Becke’s (B) [11] threeparameter hybrid model, Lee, Yang and Parr’s (LYP) correlation functional [12] under Pople’s 6-31g (d,p) basis set has been employed to optimize and elucidate their thermal and molecular orbital properties [13]. Initial optimization of all compounds was performed in the gas phase. Dipole moment, electronic energy, enthalpy, free energy and electrostatic potential are calculated for all the compounds (Figure 1).
Figure 1: Chemical structure of Metronidazole (Met) and its modified analogues.

Frontier molecular orbital features HOMO (highest occupied molecular orbital), LUMO (lowest unoccupied molecular orbital) were calculated at the same level of theory. For each of the drugs, HOMO-LUMO energy gap, hardness (η), softness (S) and chemical potential were calculated from the energies of frontier HOMO and LUMO as reported considering Parr and Pearson interpretation [14,15] of DFT and Koopmans theorem [16] on the correlation of ionization potential and electron affinities with HOMO and LUMO energy (𝜀). The following equations are used to calculate hardness (η), softness (S) and chemical potential (μ);
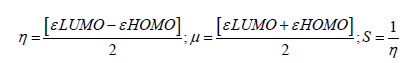
In computer aided drug discovery system, computational predictions are using to explore absorption, distribution, metabolism, excretion, and toxicity (ADMET) which saves on time and investment. AdmetSAR online database was utilized to predict ADMET properties of Metronidazole and its analogues [17].
Result and Discussion
Thermodynamic Analysis
Simple modifications of molecular structure significantly influence the structural properties including thermal and molecular orbital parameters. From the free energy, and enthalpy values, spontaneity of a reaction and stability of a product can be predicted [18]. In drug design, hydrogen bond formation and nonbonded interactions also influenced by dipole moment. Increased dipole moment can improve the binding property [19]. From thermodynamic data (Table 1), the free energy of Metronidazole is -623.7600 Hartree, where M1 shows the highest negative value (-921.4882 Hartree). The –F substitution (M1) influence the free energy significantly. Highly negative free energy is favourable for stable configuration. Again, the dipole moment of Metronidazole is 4.1174 Debye where M3 shows the maximum dipole moment (5.3559 Debye) due to substitution of –NH2 group (Figure 2).
Table 1: The stoichiometry, molecular weight, electronic energy, enthalpy, free energy in Hartree and dipole moment (Debye) of Metronidazole (Met) and its analogues.

Figure 2: Most stable optimized structures of Metronidazole (Met) and newly designed analogues. Optimized with B3LYP/6- 31g (d, p) level theory.

Molecular Orbital Properties
Table 2: Energy (eV) of HOMO, LUMO, gap, hardness, softness and chemical potential of the designed drugs.

The HOMO-LUMO energies, hardness, softness, chemical potential of all compounds is presented in Table 2. The electronic absorption relates to the transition from the ground state to the first excited state and mainly described by one electron excitation from HOMO to LUMO [20]. The chemical hardness, softness, and potential values depend on the energy gap of HOMO-LUMO [21,22]. Kinetic stability decreases with the decrease of HOMO-LUMO gap. As a result, removal of electrons from ground state HOMO to excited state LUMO requires less energy. In our studies, Metronidazole shows the HOMO-LUMO gap 4.6124 eV, where M3 have the lowest energy gap (4.1783 eV) with highest softness (0.4787 eV) which may contribute higher chemical reactivity (Figure 3).
Figure 3: Frontier molecular orbital (HOMO-LUMO) and related energy of Metronidazole (Met) and M3.

Molecular Electrostatic Potential Analysis
Molecular electrostatic potential (MEP) was calculated at B3LYP/6-31G (d,p) level of theory to forecast the reactive sites for electrophilic and nucleophilic attack of all optimized structures [23]. Red color represents maximum negative area which favorable site for electrophilic attack, blue color indicates the maximum positive area which favorable site for nucleophilic attack and green color represent zero potential area. MEP displays molecular size, shape as well as positive, negative and neutral electrostatic potential regions simultaneously in terms of color grading. It is seen from MEP map, region having the negative potential are over electronegative atom (oxygen atoms) and having positive potential are over hydrogen atoms. Here, the maximum negative potentiality is found for M3 is -0.3497 a.u (deepest red) for oxygen atoms and the highest positive potentiality of M1 is +0.3903 a.u (deepest blue) of hydrogen atoms (Figure 4).
Figure 4: Molecular electrostatic potential maps of Metronidazole (Met), M1 and M3.

ADMET Predictions
According to AdmetSAR data (Table 3), all the analogues are non-carcinogenic and show positive response for blood brain barrier (BBB) and human intestinal absorption criteria. All the structures show III category acute oral toxicity; as a result, they are safe for oral administration. All drugs are P-glycoprotein non-inhibitor where, P-glycoprotein inhibition can interrupt the absorption, permeability and retention of the drugs [24]. However, furthermore study of this aspect is necessary [24].
Table 3: Selected pharmacokinetic parameters of Metronidazole and its designed derivatives.

Here, NI= non inhibitor, NC= non carcinogenic
Conclusion
From quantum chemical calculations, newly designed analogues have improved thermodynamic properties than Metronidazole. M2 and M3 have smaller HOMO-LUMO gap with higher chemical reactivity than the parent drug. ADMET results predict that, all analogues are non-carcinogenic and safe for oral administration. Considering above investigation, this study can be helpful to design a new potent possible candidate for the better performance.
References
- Ahmed MJ, Theydan SK (2013) Microwave assisted preparation of microporous activated carbon from Siris seed pods for adsorption of metronidazole antibiotic. Chemical Engineering Journal 214: 310-318.
- Kapoor K, Chandra M, Nag D, Paliwal JK, Gupta RC, et al. (1999) Evaluation of metronidazole toxicity: a prospective study. International journal of clinical pharmacology research 19(3): 83-88.
- Andrzejewska M, Yépez Mulia L, Cedillo Rivera R, Tapia A, Vilpo l, et al. (2002) Synthesis, antiprotozoal and anticancer activity of substituted 2-trifluoromethyl-and 2-pentafluoroethylbenzimidazoles. European journal of medicinal chemistry 37(12): 973-978.
- Brogden RN, Heel RC, Speight TM, Avery GS (1978) Metronidazole in anaerobic infections: a review of its activity, pharmacokinetics and therapeutic use. Drugs 16(5): 387-417.
- Rossi S (2013) Australian Medicines Handbook (2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN 978-0-9805790- 9-3.
- Schneider G, Fechner U (2005) Computer-based de novo design of druglike molecules. Nature Reviews Drug Discovery 4(8): 649.
- Juillerat Jeanneret L, Schmitt F (2007) Chemical modification of therapeutic drugs or drug vector systems to achieve targeted therapy: looking for the grail. Medicinal research reviews 27(4): 574-590.
- Gleeson MP, Gleeson D (2009) QM/MM Calculations in Drug Discovery: A Useful Method for Studying Binding Phenomena? Journal of Chemical Information and Modeling 49(3): 670-677.
- Pence HE, Williams A (2010) ChemSpider: An Online Chemical Information Resource. Journal of Chemical Education 87(11): 1123- 1124.
- Gaussian09 RA (2009) 1, MJ Frisch, GW Trucks, HB Schlegel, GE Scuseria, MA Robb, JR Cheeseman, G. Scalmani, V. Barone, B. Mennucci, GA Petersson et al. [Eds.], Gaussian. Inc, Wallingford CT.
- Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A 38: 3098-3100.
- Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37(2): 785-789.
- Kruse H, Goerigk L, Grimme S (2012) Why the Standard B3LYP/6-31G* Model Chemistry Should Not Be Used in DFT Calculations of Molecular Thermochemistry: Understanding and Correcting the Problem. The Journal of Organic Chemistry 77(23): 10824-10834.
- Calais J L (1993) Density-functional theory of atoms and molecules. R.G. Parr and W. Yang, Oxford University Press, New York, Oxford, 1989. IX + 333 pp. Price £45.00. International Journal of Quantum Chemistry 47(1):101.
- Pearson RG (1995) The HSAB Principle-more quantitative aspects. Inorganica Chimica Acta 240(1-2): 93-98.
- Pearson RG (1986) Absolute electronegativity and hardness correlated with molecular orbital theory. Proceedings of the National Academy of Sciences 83(22): 8440-8441.
- Cheng F, Li W, Zhou Y, Shen J, Wu Z, et al. (2012) admetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties. Journal of Chemical Information and Modeling 52(11): 3099-3105.
- Cohen N, Benson SW (1993) Estimation of heats of formation of organic compounds by additivity methods. Chemical Reviews 93(7): 2419-2438.
- Lien EJ, Guo ZR, Li RL, Su CT (1982) Use of dipole moment as a parameter in drug-receptor interaction and quantitative structure-activity relationship studies. Journal of Pharmaceutical Sciences 71(6): 641-655.
- Saravanan S, Balachandran V (2014) Quantum chemical studies, natural bond orbital analysis and thermodynamic function of 2, 5-dichlorophenylisocyanate. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 120: 351-364.
- Azam F, Alabdullah NH, Ehmedat HM, Abulifa AR, Taban I, et al. (2018) NSAIDs as potential treatment option for preventing amyloid β toxicity in Alzheimer’s disease: an investigation by docking, molecular dynamics, and DFT studies. Journal of Biomolecular Structure and Dynamics 36(8): 2099-2117.
- Parr RG, Zhou Z (1993) Absolute hardness: unifying concept for identifying shells and subshells in nuclei, atoms, molecules, and metallic clusters. Accounts of Chemical Research 26(5): 256-258.
- Politzer P, Murray JS (1991) Molecular electrostatic potentials and chemical reactivity. Reviews in computational chemistry 2: 273-312.
- Amin ML (2013) P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights 7: 27-34.

Editorial Manager:
Email:
biomedicalengineering@lupinepublishers.com

-

Mark E Smith
Bio chemistry
University of Texas Medical Branch, USA -

Lawrence A Presley
Department of Criminal Justice
Liberty University, USA -

Thomas W Miller
Department of Psychiatry
University of Kentucky, USA -

Gjumrakch Aliev
Department of Medicine
Gally International Biomedical Research & Consulting LLC, USA -

Christopher Bryant
Department of Urbanisation and Agricultural
Montreal university, USA -

Robert William Frare
Oral & Maxillofacial Pathology
New York University, USA -

Rudolph Modesto Navari
Gastroenterology and Hepatology
University of Alabama, UK -

Andrew Hague
Department of Medicine
Universities of Bradford, UK -

George Gregory Buttigieg
Maltese College of Obstetrics and Gynaecology, Europe -

Chen-Hsiung Yeh
Oncology
Circulogene Theranostics, England -
.png)
Emilio Bucio-Carrillo
Radiation Chemistry
National University of Mexico, USA -
.jpg)
Casey J Grenier
Analytical Chemistry
Wentworth Institute of Technology, USA -

Hany Atalah
Minimally Invasive Surgery
Mercer University school of Medicine, USA -

Abu-Hussein Muhamad
Pediatric Dentistry
University of Athens , Greece
