




Lupine Publishers Group
Lupine Publishers
ISSN: 2637-4609
Review Article(ISSN: 2637-4609) 
Computational modelling Procedures for Geometry Optimization, Kinetic and Thermodynamic Calculations using Spartan Software - A Review Volume 1 - Issue 5
Received: February 02, 2018; Published: February 21, 2018
Corresponding author: Adeboye Omolara, Chemistry Department, Emmanuel Alayande College of Education, P. M. B 1010, Nigeria
DOI: 10.32474/AOICS.2018.01.000123
Also view in:




Abstract
Computational chemistry is a technique used only by people that have experience in using tools that are difficult to understand and apply. This reviewed article give insight to simple procedure that can be used to optimize geometry of molecules to obtain geometric parameters such as bond length, bond angles and dihedral angles and also to carry out kinetic and thermo dynamic calculations on uni- molecular reactions involving a first order and bimolecular reactions involving second order reactions.
Keywords: Procedures; Optimization; Kinetics; Spartan; Thermodynamics
Introduction
Today, advances in software have produced programs that are easily used by chemists. Along with new software comes new literature on the subject. Different books are available to describe the fundamental principles of computational chemistry at almost every level of detail. A number of books also exist that explain how to apply computational chemistry techniques to simple calculations appropriate for student assignments [1]. However, there is dearth of information on detailed and simple progressive procedure for calculations using Spartan software, hence this review.
Conformer Calculation
Conformational search was done using molecular mechanics force field (MMFF) to obtain the most stable conformer. The most stable structure is the structure that has the lowest energy. Draw the structure. From Calculation: Calculate click Conformer distribution from the menu list with Molecular Mechanics MMFF. Click Global calculations and Click submit
Reaction Path Calculation (For Kinetic Study)
Reaction path study is important to obtain a clear insight into the true and correct transition state so as to obtain correct kinetic and thermodynamic values. This was carried out by selecting reaction coordinates for the molecule or the reaction, here the internal coordinates would be varied from its initial bond distance in the stable reactant form to its bond distance in the product form values in the product form and set calculation into desired iterations (2030 steps of calculations). Set the transition State Structure select the reaction coordinate. Set the bond distance in the reactant form and set the bond distance in the product form. Select Energy Profile in calculate. Click Submit. Open the result and click to animate the different Structures and the corresponding Energies of formation. The first structure has the Lowest Energy and it is the most Stable Structure.
Geometry Optimization
Geometry optimization is very important in Computational Chemistry. It involves taking the geometric approximations and making them as exact as possible. It involves a series of computing process or repeated mathematical calculations performed on the molecule until the energy of the molecule reach a minimum. Three types of calculations can be performed:
a) Molecular Mechanics
b) Ab-Initio
c) Semi-Empirical Calculation
These Calculations are used to optimize the geometry and this depends basically on the type of level of calculations and the basis set used. The geometry of a molecule determines the physical and chemical properties so to have accurate molecular geometry is very important. Three measurements that are very important in calculating the molecular geometry are:
i Bond Angles (degrees)
ii. Bond Distance (Angstrom)
iii. Dihedral Angles (degrees)
Molecular geometry is simply the molecule's arrangement of atoms in space. This arrangement and the electrons around the atom determine the energy level of that molecule. The energy of a molecular system varies even when there is little change in its structure. The main aim of geometry optimization is to determine the atomic arrangement that makes a molecule stable. Molecules are most stable and most likely to be found in nature at very low energy. In order to optimize a molecular geometry, one would want to test various possibilities to see which will give the lowest energy value. This is achieved by creating a Potential Energy Surface (PES). PES is the mathematical expression that shows the relationship between the different molecular geometries and their corresponding single - point energies.
Single Point Energy Calculation
This is performed to obtain the correct molecular structure with the lowest energy value.
Click new from file on the worksheet of spartan. build a molecule of ethyl acetate for example using appropriate hybridization in the model kit. choose geometry and click constrain dihedral
Click On the C, O, C, C Atoms (Figure 1)
Figure 1:

Click on the pink constrain marker that appears on the C-O Bond
Choose display click properties
Click on the dynamic box in the constrain properties window. Change the values to read 180.000 to 0.000 and change steps to 20. Hit the enter key
Choose Set up, click calculation select energy profile at the ground state with semi - empirical pm3 or any other levels of calculations.
Click submit and save as ethylacetate spartan.
Click ok to started calculation and click ok to completed calculation.
Click yes to open all the possible molecular structures.
Choose display. Click spreadsheet click on the box with grey shade click add select rel. energy.
Click ok.
To add angles to the spreadsheet,
Choose geometry click measure dihedral.
Click on the constrain marker on the c - o bond.
Click on the yellow p box on the right of the screen Choose display click plot
Choose X axis click dihedral and Y axis click e (kj/mol).
Click Ok.
Press play to view the animation of the bond rotation.
Save and Close The File.
Geometry Optimization
Click New from file to display the worksheet
Use the model kit to build a molecule using appropriate hybridizations to build ethyl acetate (Figure 2).
Figure 2:

Choose the structure with the lowest minimization energy value.
To minimize the energy click on the icon E the energy value is shown at the lower part of the workspace.
Choose set up to calculate the equilibrium geometry at the ground state with semi empirical PM3 or other calculation methods.
Start from current geometry click orbitals and energies and charges and bond order on print.
Set Options OPTCYCLE=200-5000 in case the calculation ran out of cycle.
Click Converge
Click Submit
Save as ethylacetate. Spartan in a folder.
Click ok to start the calculation Click ok to completed calculation Click display
Select output to read the energy of formation and other parameters
Choose display to display the output, properties, orbiral energies (homo-lumo), spectra, formulas.
Click on the icon ? Measure distance to determine the bond length.
Click on the icon <? Measure angleto determine the bond angle
Click on the icon'?'Measure dihedral
Record the values for the optimized structure.
Predicting The Transition State Structure
Click New from file.
Build a molecule using the appropriate hybridization from the model kit.
Click on E to minimize the Energy of the structure
Record the minimization energy.
Click View to display the Structure on the Screen.
Choose geometry and click constrain bond distance from the predicted transition state bond distance to the normal bond distance (path studies).
Click the dynamic box to set the values.
Hit the enter key
Click set up to choose calculation
Select energy profile at the ground state with semi empirical pm3
Click Submit and Save.
Click ok to start calculation
Click ok to completed calculation
Click yes to open all the possible molecular structures.
Click ok
Press Play to view the animation of the bond breaking and bond formation.
Transition State Geometry Optimization
Build the structure with proper orientation and mechanism using the model kit.
Minimize the energy using the icon E.
Click on the icon to guess transition state and show the suggested mechanism of the reaction.
Click on a double arrow head at the lower right corner of the worksheet to display the Transition State Structure and possibly to confirm the mechanism.
Click set up to calculate the transition state geometry at the ground state with semi empirical PM3 and other calculation methods.
Select compute ir, orbital energies, thermodynamics, vibrational modes, charges and bond order on print.
Click options optcycle =250
Click converge
Click submit
Click ok to started calculation
Click ok to completed calculation
Select output to read the heat of formation and other parameters [2-4]
Kinetic and Thermodynamic Calculations
The kinetic and thermodynamic parameters were obtained using the sum of the ground state energy (GSE) and the statistical mechanically calculated enthalpy to arrive at a closer approximation of the true energy of the molecule. The enthalpy of a species will be defined as:
Hi = GSEi + Hism
Where the superscript 'sm' is the statistical mechanically calculated enthalpy. Substitute this into the initial definition of the heat of reaction we have:
ΔHrxn = (GSEproduct + Hsm product)- (GSEreactant + Hsm reactnt)
Activation energy (Ea) was calculated according to the transition state theory for a unimolecular reaction at 698K [2,5].
Ea = ΔH * - RT
The entropy of the reaction was calculated by taking the difference of product and reactant entropies that is
ΔSreaction = SProduct - Sreactants
And ΔSactivated=Stransition- Sreactant
The Gibbs free energy and Gibbs free energy of activation was calculated using the modified version of the heat of reaction equation. Knowing that
G = H - TS and ΔG = ΔH * - TΔS
The first order coefficient k(T) was calculated using transition state theory (TST) [6] assuming that the transmission coefficient is unity as shown in the following equation
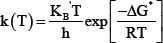
Where ΔG^* is the Gibbs free energy change between the reactant and the transition state and K^'and h are the Boltzmann and Planck constants respectively.
Pre -exponential factor is given as 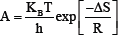
Arrhenius rate was obtained using the rate equation [7]
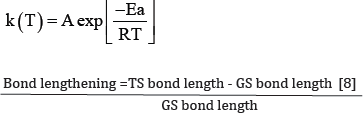
The bond lengthening character was used to describe the extent of bond lengthening to show the rate determining step and to explain the concertedness and synchronicity of the reaction.
Conclusion
This detailed and simple progressive procedure for calculations described using Spartan software has been used for computational modeling of kinetic and thermodynamic [9-16] studies of various reactions.
References
- David C Young (2001) Computational Chemistry: A Practical Guide for Applying Techniques to Real-World Problems. John Wiley & Sons Inc, England.
- J Warren Hehre (2003) A Guide to Molecular Mechanics and Quantum Mechanical Chemical Calculations Irvine, USA PP 399.
- Warren Hehre and Sean Ohlinger (2009) Spartan 06 for Windows, Machintosh and Linux tutorial and user's guide. Wavefunction Inc, USA.
- Warren Hehre and Sean Ohlinger (2010) Spartan 10 for Windows, Machintosh and Linux tutorial and user's guide. Wavefunction Inc, USA.
- E V Anslyn and DA Dougherty (2006) Modern Physical Organic chemistry 2006 University Science Books. Wilsted and Taylor Publishing Services USA.
- S W Benson, O Neal (1970) Kinetic Data of Gas Phase Unimolecular Reactions. NSRDS- MBS 21.
- M R Wright (2004) An Introduction to Chemical Kinetics. John Wiley &Sons, England.
- L Hamon, J Levaisalles, YL Pascal (1989) A non-empirical 1,5 elimination transition structure for ethyl formate. Tetrahedron 45: 1711-1722. Robinson, KA Holbrook (1972) Unimolecular Reactions. John Wiley & Sons Ltd, England.
- Omolara Olubunmi Adeboye (2013) Quantum Mechanical Studies of the Kinetics, Mechanisms and Thermodynamics of the Gas-Phase Thermal Decomposition of Ethylvinyl Ether. Southern Brazilian Journal of Chemistry 21(21): 41-60.
- Adejoro IA, Adeboye OO, Esan T (2013) Ab-Initio and DFT Studies of the Kinetics, Mechanisms and Thermodynamics of the Gas- Phase Pyrolysis of Ethyl Bromide. African Journal of Pure and Applied Chemistry 7(6): 231-241.
- Adeboye OO (2014) Quantum Mechanical Study of the Kinetics, Mechanisms and Thermodynamics of the Gas-Phase Pyrolysis of Isopropyl Bromide. IISTE Chemistry and Material Research 6(10): 1-9.
- Adejoro Isaiah Ajibade, Adeboye Omolara Olubunmi (2016) Computational studies on the a- and p- Elimination Pathways for the Kinetics and Thermodynamics of the Gas-Phase Pyrolysis of Allyl Formates. Nature and Science: 14(4): 74-82.
- Adejoro I A, Esan TO, Adeboye OO, Adeleke BB (2016) Quantum Mechanical Studies of the Kinetics, Mechanisms and Thermodynamics of the gas-phase Thermal Decomposition of Ethyl Dithiocarbonate (Xanthate). Journal of Taibah, University of Science, Elsiever.
- Adeboye OO (2017) Computational Modelling of the Mechanisms, Kinetics and Thermodynamics of Pyrolysis of Isobutyl Bromide in the Gas-Phase. Chemical Science International Journal 18(4): 1-9.
- Adeboye OO (2017) The Mechanisms, Kinetics and Thermodynamics of the Gas-Phase Pyrolysis of sec-Butyl Bromide: A Computational Approach. International Journal of Pure and Applied Chemistry: 14 (2).



-

Mark E Smith
Bio chemistry
University of Texas Medical Branch, USA -

Lawrence A Presley
Department of Criminal Justice
Liberty University, USA -

Thomas W Miller
Department of Psychiatry
University of Kentucky, USA -

Gjumrakch Aliev
Department of Medicine
Gally International Biomedical Research & Consulting LLC, USA -

Christopher Bryant
Department of Urbanisation and Agricultural
Montreal university, USA -

Robert William Frare
Oral & Maxillofacial Pathology
New York University, USA -

Rudolph Modesto Navari
Gastroenterology and Hepatology
University of Alabama, UK -

Andrew Hague
Department of Medicine
Universities of Bradford, UK -

George Gregory Buttigieg
Maltese College of Obstetrics and Gynaecology, Europe -

Chen-Hsiung Yeh
Oncology
Circulogene Theranostics, England -
.png)
Emilio Bucio-Carrillo
Radiation Chemistry
National University of Mexico, USA -
.jpg)
Casey J Grenier
Analytical Chemistry
Wentworth Institute of Technology, USA -

Hany Atalah
Minimally Invasive Surgery
Mercer University school of Medicine, USA -

Abu-Hussein Muhamad
Pediatric Dentistry
University of Athens , Greece
